
-
Argentina

-
Australia

-
Austria

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil

-
Canada (English)

-
Canada (Français)
-
Chile

-
Colombia
-
Croatia

-
Denmark

-
Deutschland

-
Europe

-
France

-
Greece (Ελληνικά)

-
Italia

-
Hungary

-
Lietuva

-
México

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland

-
Norge

-
Oman (العربية)

-
Oman (English)
-
Polska

-
Portugal

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain

-
Suomi

-
Sverige

-
Schweiz

-
台灣 (中文)

-
United States

-
UAE (العربية)

-
UAE (English)

 Sobre la AME
Sobre la AME
Introducción a la Atrofia Muscular Espinal (AME)
Los primeros pasos para entender la Atrofia Muscular Espinal (AME).
La Atrofia Muscular Espinal (AME) es una enfermedad rara causada por alteraciones genéticas que afectan a las células conocidas como neuronas, que controlan diversos músculos del cuerpo. Por lo tanto, la AME es un padecimiento genético y neuromuscular progresivo que según la clasificación conduce a diferentes niveles de severidad que impacta en la calidad de vida del paciente1.
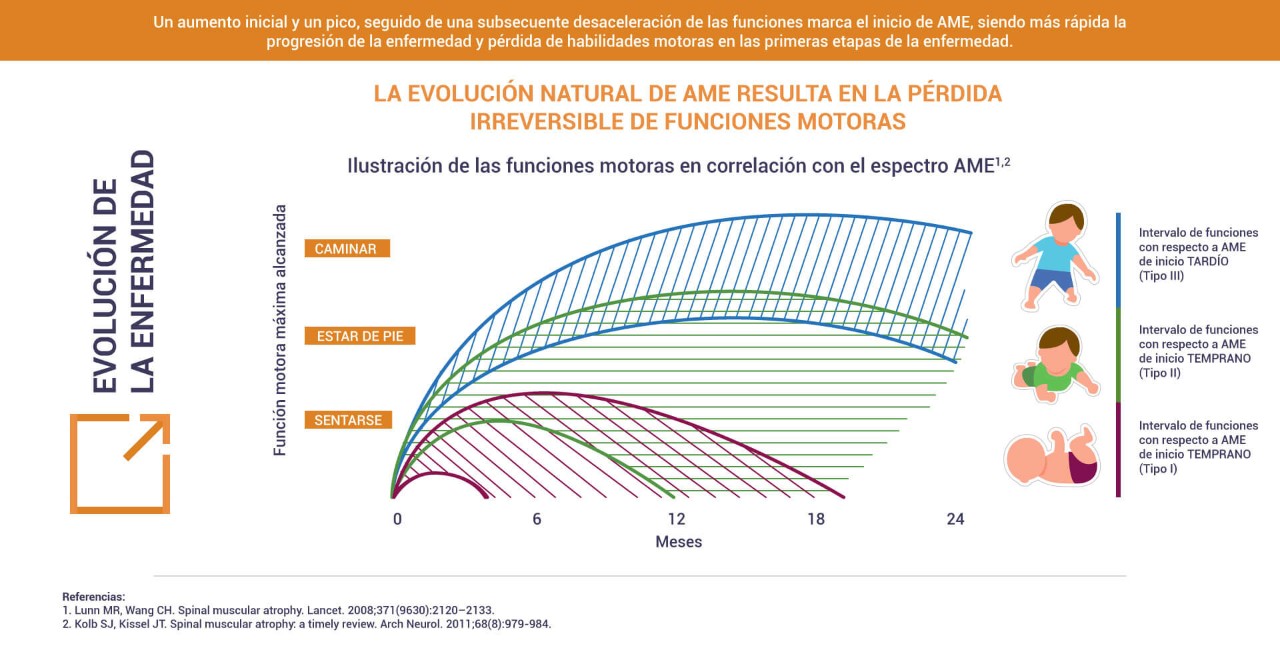
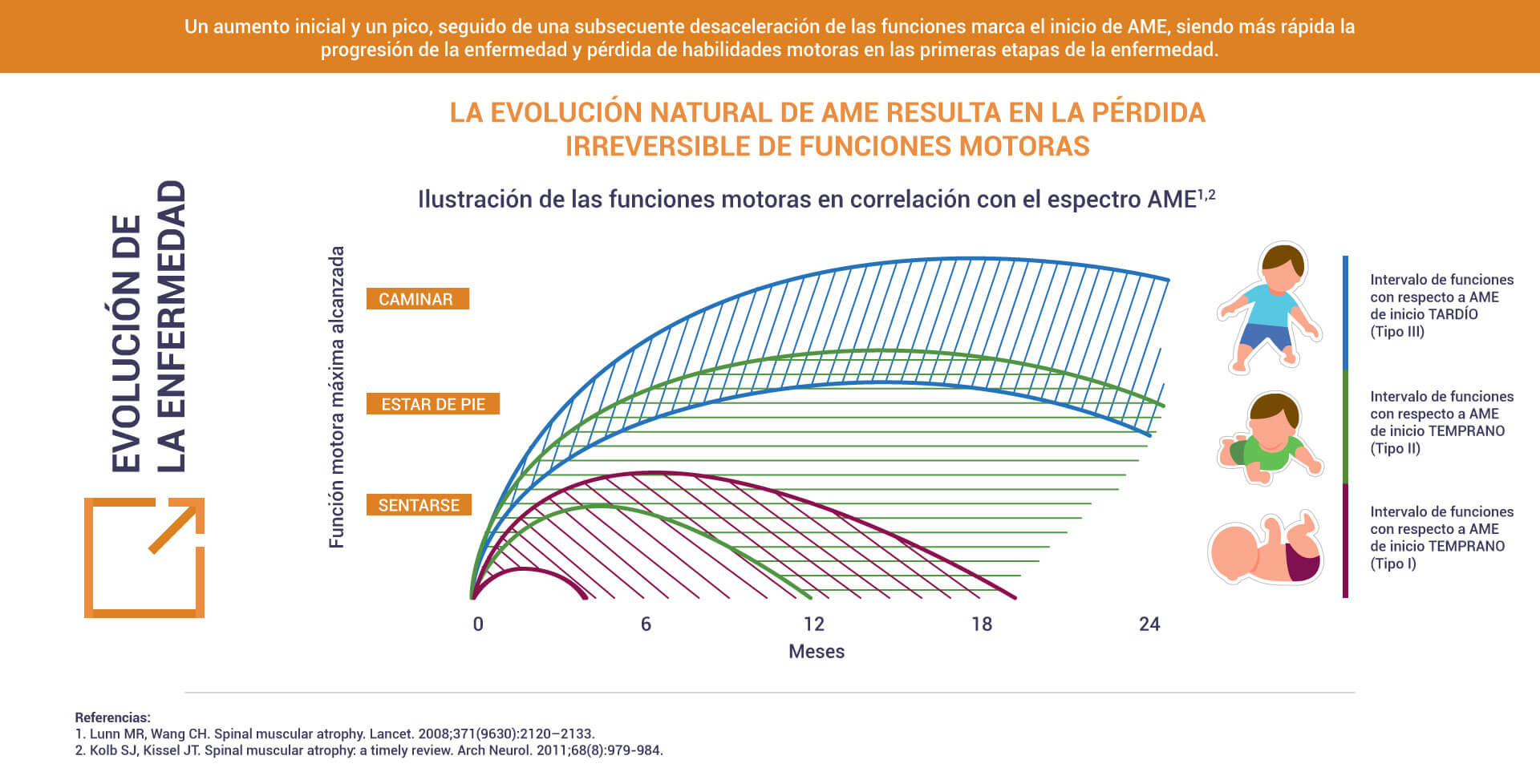
La AME fue descrita por primera vez hace casi 130 años2. Desde entonces, se han desarrollado varios estudios que han aumentado considerablemente el conocimiento sobre su causa.
Origen de la Atrofia Muscular Espinal (AME)
Hoy en día, se sabe que es originada por cambios en el gen SMN1, localizado en el cromosoma 5q, de ahí el nombre AME 5q. Este gen es responsable de la producción de la proteína de supervivencia de la neurona motora, conocida como SMN3.


Hay otros tipos de AME causadas por diferentes genes, conocidas como AME no-5q4. En este sitio web sólo trataremos la AME 5q, que se llamará sólo AME, para una mayor fluidez de lectura.
La correlación entre el número de copias de SMN2 y las manifestaciones clínicas de la Atrofia Muscular Espinal (AME).
La supervivencia de las neuronas motoras depende de la cantidad y calidad de la proteína SMN, la cual es producida por el gen SMN1.
En los pacientes con AME, que no producen la proteína SMN funcional del gen SMN1, el gen SMN2 reemplaza parcialmente esta producción. Aun así, la cantidad de proteína funcional producida por el gen SMN2 no es suficiente para mantener la supervivencia de las neuronas motoras y evitar la manifestación de la enfermedad14.
A diferencia de la mayoría de los genes, que suelen estar presentes en dos copias (una del padre y otra de la madre), el gen SMN2 puede tener un número bastante variable de copias. Por lo tanto, la cantidad de proteína SMN funcional producida por el gen SMN2 también será variable.
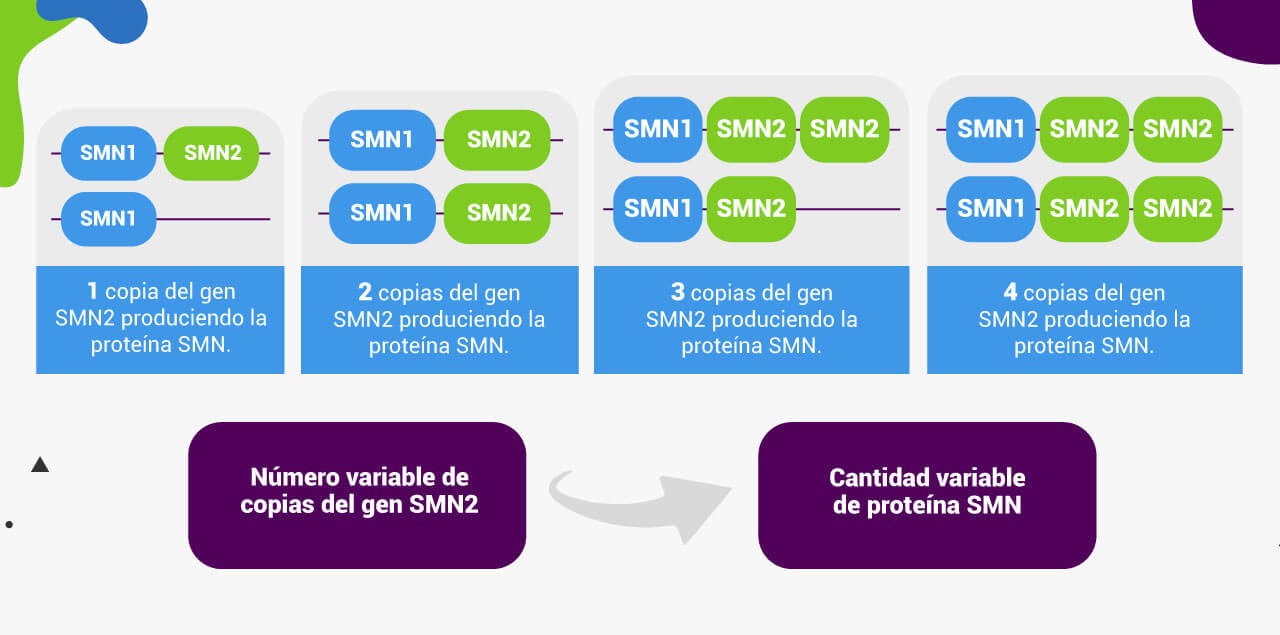
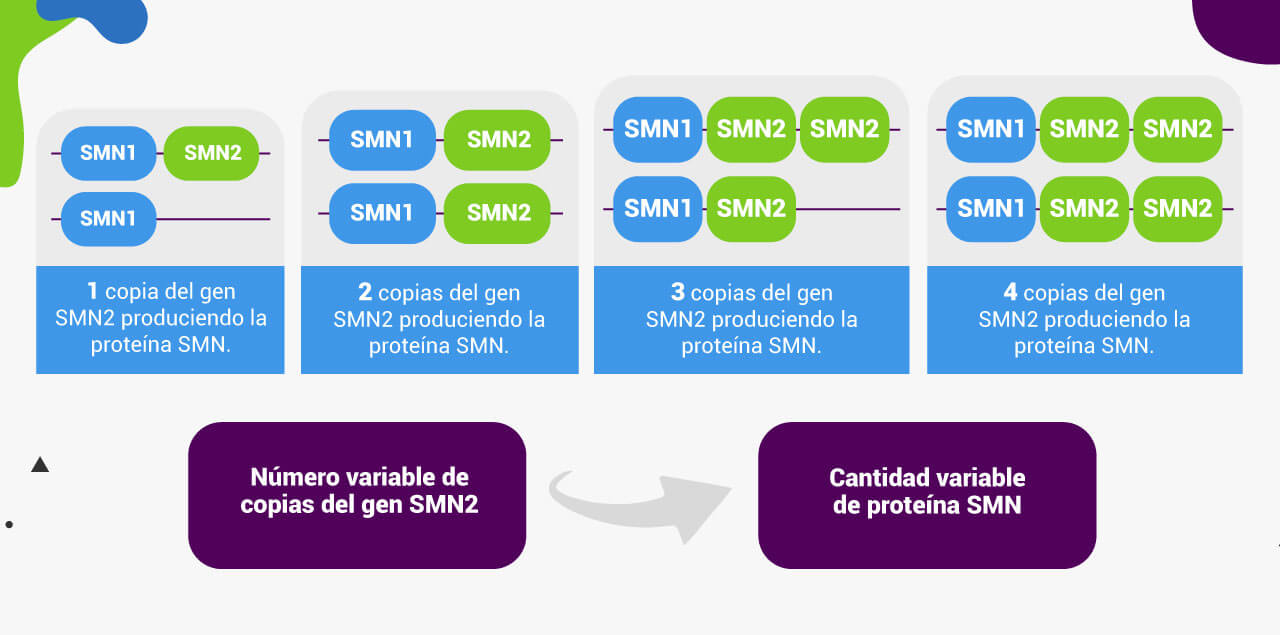
El siguiente gráfico muestra información sobre el tipo de AME y el número de copias de SMN2 de aproximadamente 3,500 pacientes. Se puede observar que existe una correlación entre el número de copias de SMN2 y el tipo de AME.
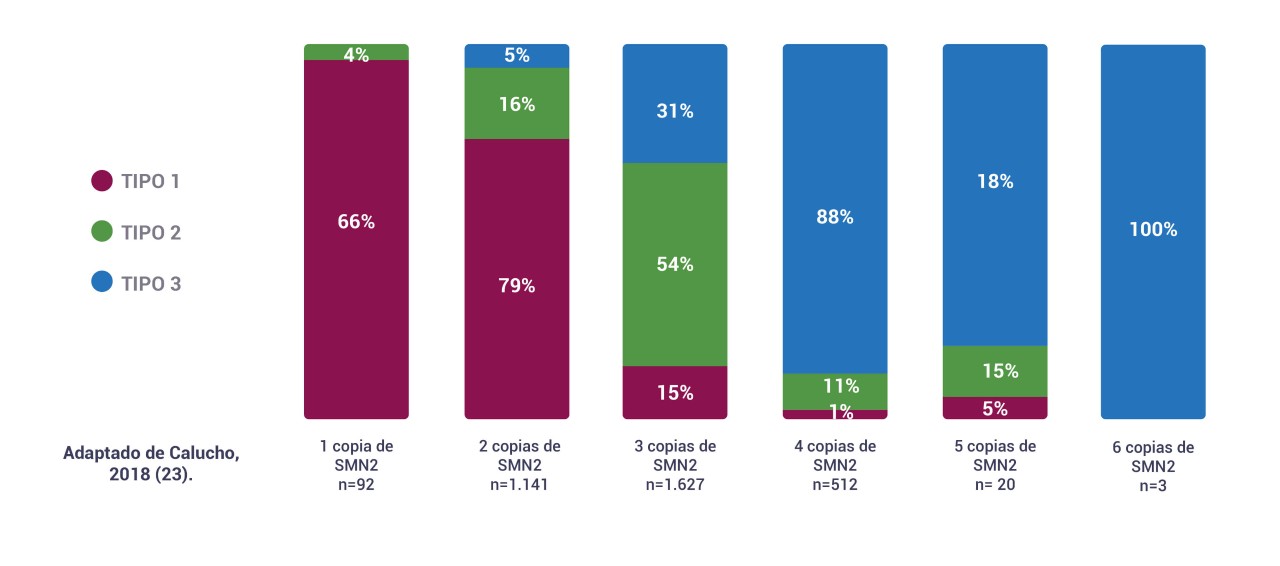
Sin embargo, la correlación anterior no es absoluta ya que existen otros factores modificadores de la severidad de la enfermedad, por lo que es importante destacar que el número de copias del gen SMN2 no se puede utilizar para determinar por completo el pronóstico de los pacientes14.
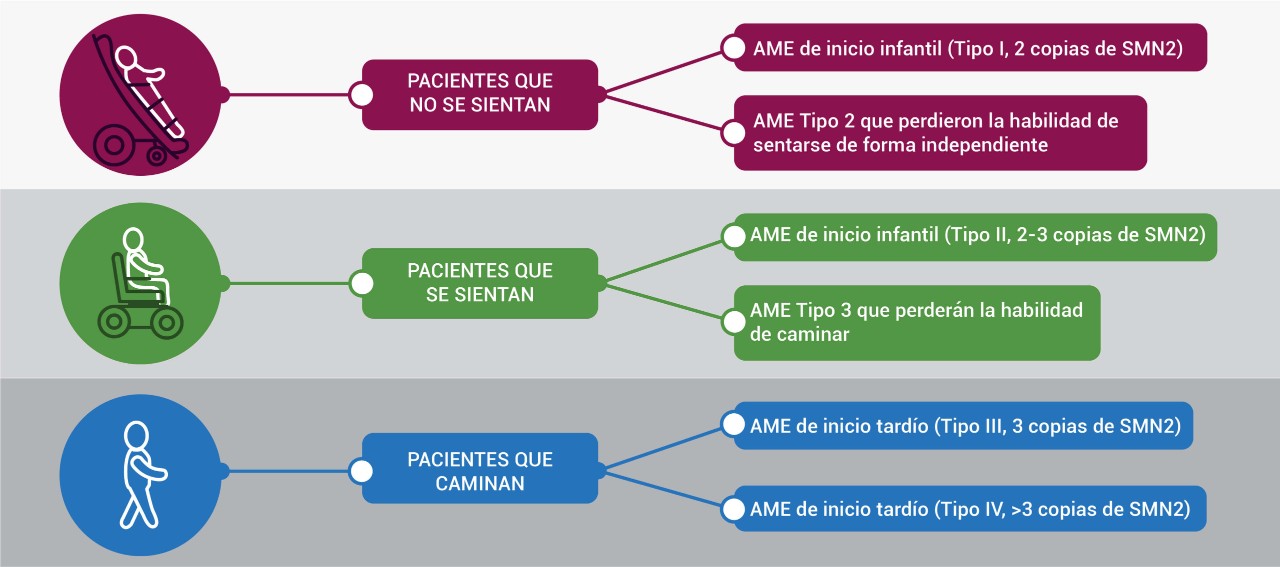
.
Referencias
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630): 2120–2133.
2. Kolb SJ, Kissel JT. Spinal muscular atrophy: a timely review. Arch Neurol. 2011;68(8):979-984.
3 Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65.
4. Farrar MA, Kiernan MC. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics. 2015;12(2):290–302.
5. Rouault F, Christie-Brown V, Broekgaarden R, et al. Disease impact on general well-being and therapeutic expectations of European Type II and Type III spinal muscular atrophy patients. Neuromuscul Disord. 2017;27(5):428-438.
6. Hunter M, Heatwole C, Luebbe E, Johnson NE. What Matters Most: A Perspective From Adult Spinal Muscular Atrophy Patients. J Neuromuscul Dis. 2016;3(3):425-429.
7. Qian Y, McGraw S, Henne J, et al. Understanding the experiences and needs of individuals with Spinal Muscular Atrophy and their parents: a qualitative study. BMC Neurol. 2015;15:217.
8. Jeppesen J, Madsen A, Marquardt J, Rahbek J. Living and ageing with spinal muscular atrophy type 2: observations among an unexplored patient population. Dev Neurorehabil. 2010;13(1):10-18.
9 Higgs EJ, McClaren BJ, Sahhar MAR, et al. "A short time but a lovely little short time": Bereaved parents' experiences of having a child with spinal muscular atrophy type 1. J Paediatr Child Health. 2016;52(1):40-46.
10. Laufersweiler-Plass C, Rudnik-Schöneborn S, Zerres K, et al. Behavioural problems in children and adolescents with spinal muscular atrophy and their siblings. Dev Med Child Neurol. 2003;45(1):44-49.
11. Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-115.
12. Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018;28(3):197-207.
13. El contenido discutido se inspiró en la Guía de Discusión sobre la Atrofia Muscular Espinal (AME) en Brasil: Trabajando hoy para cambiar mañana. Haga clic para descargar la Guía de forma gratuita y disponible en su totalidad.
14. Calucho M, Bernal S, Alías L, et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28(3):208-215.
¿Tienes dudas sobre algún término de este artículo? Consulta el glosario.
#AtrofiaMuscularEspinal #AME #EnfermedadRara